Questionnaire
- Advertisements
- Biological Safety
- Confidentiality
- Consent Procedures
- Design
- Device Information
- Drug/Substance/Nutrient/Biologic
- Emergency Use
- Exempt Category
- Expedited Review
- Financial Considerations
- Funding Source
- HIPAA
- Human Subject Research
- Humanitarian Use Devices
- Locations of Research
- Potential Benefits
- Radiation Safety
- Risk Assessment
- Sample Collection
- Subjects
Watch Video
- Protocol Training Video
Click arrows in the upper-left corner of video to navigate by section.
Reference Guides
- It is important to select the correct protocol type on the Protocol page; otherwise, users may provide too much or not enough information about a protocol on the Questionnaire page. If you need assistance determining the type of protocol you are submitting, please review the Human Subject Regulations Decision Charts.
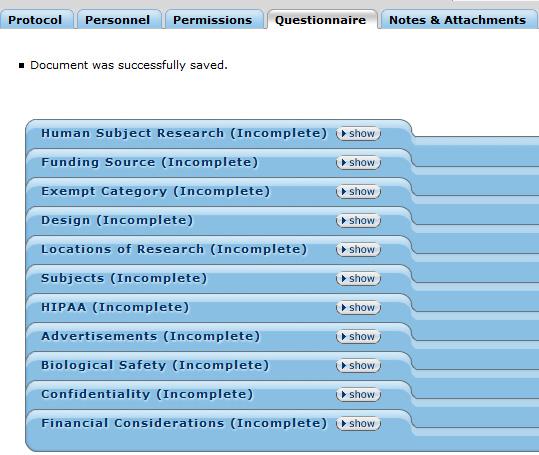
Figure 158 Protocol Document, Questionnaire Page
The Questionnaire page of the protocol document displays categorized, expandable sections that record compliance information that must be collected at state, local, and institutional levels. The save ![]() button at the bottom of the page enables users to save answer selections and entries.
button at the bottom of the page enables users to save answer selections and entries.
Questionnaire Sections/Answering Questions
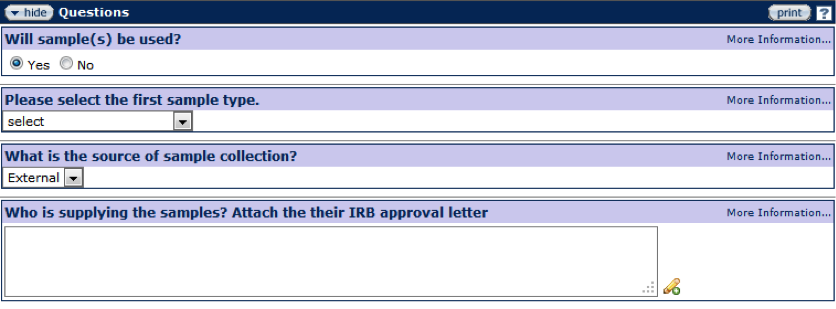
Figure 159 Protocol Document, Questionnaire Page –Section Example
Standard selection and entry tools for questions include radio ![]() buttons for selection of options for Yes/No questions and a drop-down
buttons for selection of options for Yes/No questions and a drop-down ![]() menu for multiple choice selections. For questions that require a text answer, click in the text box and enter text or click on the add note
menu for multiple choice selections. For questions that require a text answer, click in the text box and enter text or click on the add note ![]() icon to view/edit/paste text in a new browser window. Click the save
icon to view/edit/paste text in a new browser window. Click the save ![]() button at the bottom of the page to save answer selections and entries.
button at the bottom of the page to save answer selections and entries.
- Text box fields have a 2,000 character limit. WVU+kc will not give an error if the user pastes more than 2,000 characters, it will merely cut off the text that is over 2,000. If pasting a significant amount of text into a text box, double-check that all text was accepted before moving on to another question. If an answer to a question will exceed 2,000 characters, attach a separate document on the Notes & Attachments page and make note of the attachment (including the attached file name) in the answer field.
- Copying text from a Word document and pasting into a WVU+kc text box can be done; however, the text boxes in WVU+kc do not accept special formatting such as bold, italics, or bullet points. If copying from a Word document, paste into Notepad first to remove any special characters and formatting. Then, copy and paste into WVU+kc.
Answers to particular questions can cause additional questions to appear. Such logic is driven by business rules embedded into the system so that an answer given to a previous question will dynamically add one or more new questions to a section within the Questionnaire page.
- For more information about using the standard online form features to answer questions, see Selection, Entry, & Command Tools in the User Interface Orientation section.
Expand/Hide Sections
In order to display and answer questions within an individual section, click on the ![]() button; this action will only expand the section selected. Questions located within sections that are not needed will still display, but the ability to answer those questions will not be provided. After answering all questions in a required section, save
button; this action will only expand the section selected. Questions located within sections that are not needed will still display, but the ability to answer those questions will not be provided. After answering all questions in a required section, save  and collapse the individual section by clicking on the
and collapse the individual section by clicking on the ![]() button; this action will only collapse the section selected. The expand all
button; this action will only collapse the section selected. The expand all ![]() button will expand all sections on the Questionnaire page. All questions will be shown, even if they are not required. The collapse all
button will expand all sections on the Questionnaire page. All questions will be shown, even if they are not required. The collapse all ![]() button will compress all sections on the Questionnaire page.
button will compress all sections on the Questionnaire page.
Additional Information
If unsure about what a specific question is asking, click on the question mark ![]() to open the help document. This document will provide additional information about the questions being asked as well as active links to forms referenced in the questions.
to open the help document. This document will provide additional information about the questions being asked as well as active links to forms referenced in the questions.
Updates to Questionnaire
From time to time, the Questionnaire page may require updates to address new compliance/regulatory issues. If submitting an amendment or renewal to a protocol that has an older version of the questionnaire, you will be presented with the option to (1) update the questionnaire and save your answers or to (2) update the questionnaire without saving your current answers. The majority will select to copy the answers; however, if you are aware that information in a particular section will need to be significantly updated, you may want to select Do Not Copy answers in order to start new with that section. Click the update button for each section requiring an update and your questionnaire will be updated accordingly. Updates to questionnaires must be completed before you can submit an amendment or renewal.
Saving
Click the save button at the bottom of the page to save answer selections and entries. Clicking on the save button at the bottom of the page will also save an incomplete Questionnaire and allow users to return to finish it at a later date. Users will have the ability to modify previous responses as desired within a saved protocol document that has not been submitted for review.
- The act of saving the Questionnaire page does not validate the data provided; it merely saves the information provided. Please refer to the individual section title which indicates that all sections have been successfully completed before moving on to another page in the protocol document.
Session Expiration
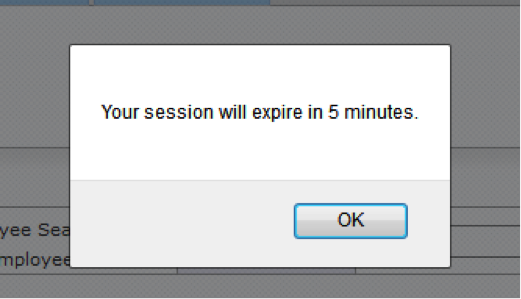
Figure 18 Protocol Document, Questionnaire Page – Session Expiration
The WVU+kc system automatically ends sessions that have been inactive for 30 minutes and will notify users that the session will expire in five minutes. It is possible to take longer than 30 minutes to complete the entire Questionnaire page; therefore, it is recommended to the protocol document after completion of each section within the Questionnaire page. If the session does expire, information will not be saved. Users will need to login to the system again in order to access the protocol from its last save or begin a new one.
- Clicking OK on the session expiration notice does not save the protocol. To continue a current session, a user must click OK and then save the protocol by clicking the save button at the bottom, center of the Questionnaire page.
Printing
To print an individual section, click the print ![]() button in the upper, right of the section label. A .pdf file of the section will populate. Users can either print the file or save to a computer.
button in the upper, right of the section label. A .pdf file of the section will populate. Users can either print the file or save to a computer.
Subtopics:
- Advertisements
- Biological Safety
- Confidentiality
- Consent Procedures
- Design
- Device Information
- Drug/Substance/Nutrient/Biologic
- Emergency Use
- Exempt Category
- Expedited Review
- Financial Considerations
- Funding Source
- HIPAA
- Human Subject Research
- Humanitarian Use Devices
- Locations of Research
- Potential Benefits
- Radiation Safety
- Risk Assessment
- Sample Collection
- Subjects
Human Subject Research
Quick Reference Guides
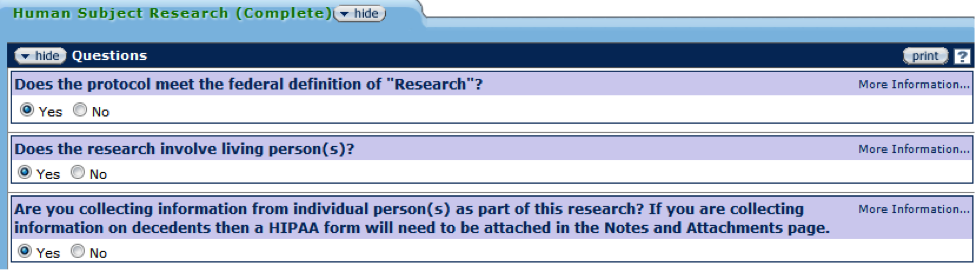
Figure 162 Protocol Document, Questionnaire Page – Human Subject Research Section
The Human Subject Research section of the Questionnaire will determine if the proposed protocol involves human subject research. Using the Human Subject Regulations Decision Charts, the responsibility for initial determination as to whether an activity constitutes human subjects research rests with the Principal Investigator and should be based on the following definitions of “research” and “human subject.” The Common Rule defines research as a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalized knowledge. Human subjects research means any activity that meets the definition of research and involves human subjects as defined by either the Common Rule or FDA regulations.
Not Human Subject Research
Projects that either do not involve human subjects or fail to meet the definition of research are excluded from IRB review. If a user answers “yes” to all three questions located within the Human Subject Research section, generally, such a project would not meet the guidelines to be considered human subject research.
- If a user answers “no” to any of the questions located within this section, and did not select NHSR as the protocol type on the Protocol page, contact the ORIC immediately by phone at 304-293-7073 or email to discuss the proposed project before continuing on to additional sections of the Questionnaire page. Determinations regarding activities that are either clearly are or clearly are not human subject research will be made by the ORIC. Determinations that are less-clear will be referred to the IRB Chair, who may make the determination or refer the matter to the full IRB.
- For more information about Human Subject Research, see Federal Policy for the Protection of Human Subjects also known as the Common Rule, view the Office for Human Research Protections (ORHP) Human Subject Regulations Decision Charts, or review the WVU ORIC NHSR Checklist.
back to top ↑
Funding Source
Figure 163 Protocol Document, Questionnaire Page – Funding Source Section
Include information about the funding source(s) for the research study in the Funding Source section of the Questionnaire. WVU charges for IRB review of all projects that are supported by an industry sponsor. If funding is provided by a public or non-profit entity, provide the name, contact person, billing address, and phone number for the funding source.
back to top ↑
Locations of Research
Watch Video
Figure 164 Protocol Document, Questionnaire Page – Locations of Research Section
From the drop-down ![]() menu of site locations, select all the sites where research will be conducted for the proposed study. If a particular location does not appear on the drop-down list, select “Off WVU Campus.” There are nine (9) location types from which to select, which are described as follows:
menu of site locations, select all the sites where research will be conducted for the proposed study. If a particular location does not appear on the drop-down list, select “Off WVU Campus.” There are nine (9) location types from which to select, which are described as follows:
| Location | Description |
|---|---|
| Chestnut Ridge Hospital | Operated by WVU Healthcare, Chestnut Ridge Center is a leading regional referral center for treatment of psychiatric illness and addiction for adults, adolescents, and children. CRH is affiliated with the Department of Behavioral Medicine and Psychiatry at the WVU School of Medicine and is located on the campus of the Robert C. Byrd Health Sciences Center in Morgantown, WV. |
| Mary Babb Randolph Cancer Center (MBRCC) | Operated by WVU Healthcare, MBRCC is WV’s premier cancer facility. It encompasses the Betty Puskar Breast Care Center, the Comprehensive Breast Care Program, Bonnie’s Bus, and the Osborn Hematopoietic Malignancy and Transplantation Program. The MBRCC is located on the campus of the Robert C. Byrd Health Sciences Center in Morgantown, WV. |
| Off WVU Campus | Any site not located on the WVU Downtown, Evansdale, or Health Sciences Campuses. Off WVU Campus sites include WVU-Affiliates WVU Institute of Technology, WVU Parkersburg, and Potomac State College. |
| Online | Research conducted online using the Internet or World-Wide Web. |
| Physician Office Center (POC) | Operated by WVU Healthcare, Physician Office Center (POC) houses the largest multi-specialty group practice in WV. |
| University Health Associates (UHA) and UHA-Affiliated | University Health Associates (UHA) is WV largest multi-specialty physician and dental group practice. UHA supports WVU’s School of Medicine and Dentistry in Morgantown and has satellite locations in Elkins, Harpers Ferry, Martinsburg, Ranson, Weston, and Wheeling, WV. |
| Veterans Administration Medical Center (VAMC) | Medical center that provides health care services to veterans. |
| Ruby Memorial Hospital or Other WVU Healthcare Site | Operated by WVU Healthcare, Ruby Memorial Hospital (RMH) is the largest facility in the WVU Hospitals system and is a teaching hospital and clinic for WVU’s health professions schools. Other WVU Healthcare sites include WVU Children’s Hospital, John Michael Moore Trauma Center, Eye Institute, Clark K. Sleeth Family Medicine Center, Cheat Lake Physicians, Preston County Pediatric and Internal Medicine, Heart Institute, Sleep Center, Sports Medicine Center, Urgent Care, Wound Management Center, and WVU Center for Reproductive Medicine. |
| WVU Campus | Any site located on the WVU Downtown, Evansdale, or Health Sciences Campuses as well as WVU Extension Offices. |
Off WVU Campus Sites
Figure 339 Protocol Document, Questionnaire Page – Locations of Research Section, Off Campus Locations
If you answer yes to the question “Will the research take place at an off campus location,” additional questions will populate in the Locations of Research section to gather more information about the location such as whether the site is domestic or international and the type of facility where the research will be conducted. Click in the text box to enter names of all the off-campus locations along with their addresses or click on the add note  icon to view/edit/paste text in a new browser window. All text boxes have a 2,000-character limit. If more space is required, attach an additional document on the Notes & Attachments page.
icon to view/edit/paste text in a new browser window. All text boxes have a 2,000-character limit. If more space is required, attach an additional document on the Notes & Attachments page.
If working with an international site, a letter from an official in the country must be attached asserting that the research will not affect the cultural mores of participants along with certification that all research documents, such as cover letters and surveys, are true translations.
IRB Authorization Agreement
Figure 340 Protocol Document, Questionnaire Page – Locations of Research Section, IAA Statement
If collaborating with another institution, indicate if WVU will be the IRB with oversight responsibility. If not, provide rationale why another institution should have IRB oversight. Attach the appropriate form on the Notes & Attachments to correspond with your answers.
| IAA Statement | Corresponding IAA Form |
|---|---|
| WVU IRB will be the IRB with oversight responsibility. | IAA-Individual Protocol-WVU IRB of Record |
| Another institution’s IRB will be the IRB with oversight responsibility. | IAA-Individual Protocol-Non-WVU IRB of Record |
Table 68 Protocol Document, Questionnaire Page – IRB Authorization Agreement Section, IAA Statement and Forms
Multi-Site Studies

Figure 341 Protocol Document, Questionnaire Page – Locations of Research Section, Multi-Site Studies
If this is a multi-site study and the lead investigator is at WVU, indicate how information will be managed to protect participants, addressing unanticipated problems, interim results, and protocol modifications.
back to top ↑
Exempt Category
Watch Video
Quick Reference Guides
Select one or more of the six Exempt Review Categories that apply to the proposed research by clicking the radio ![]() button beside the Yes or No located under the corresponding question. Finally, provide a written description of how the research meets the requirements of the exemption category by clicking to enter text or click on the add note icon to view/edit/paste text in a new browser window.
button beside the Yes or No located under the corresponding question. Finally, provide a written description of how the research meets the requirements of the exemption category by clicking to enter text or click on the add note icon to view/edit/paste text in a new browser window.
Exempt Research
All human subjects research conducted under the auspices of WVU must meet the criteria for either Exempt, Expedited, or Full Board review. This section of the Questionnaire will address questions specifically for Exempt review. Although all research using human subjects must be approved by WVU, exempt research does not require convened IRB review and approval. Exempt research is subject to institutional review and must be determined and approved by the IRB Chair or his/her designee.
| Category | Requirements |
|---|---|
| Exempt Review Category 1 | Research conducted in established or commonly accepted educational settings, involving normal educational practices. This includes research on educational strategies, comparison among instructional techniques, or classroom management methods. |
| Exempt Review Category 2 | Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior. A copy of the interview or survey questions MUST BE ATTACHED on the Notes & Attachments page.
|
| Exempt Review Category 3 | Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures, or observation of public behavior on elected or appointed public officials, candidates for public office, or federal statutes require without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter. |
| Exempt Review Category 4 | Research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens.
|
| Exempt Review Category 5 | Research and demonstration projects which are are designed to study, evaluate, or otherwise examine public benefit or service programs.
|
| Exempt Review Category 6 | Taste and food quality evaluation and consumer acceptance studies. |
Table 64 Protocol Document, Questionnaire Page – Exempt from IRB Review Section, Exemption Categories
Research activities not regulated by the FDA in which the involvement of human subjects will be in one or more of the following six categories are exempt from IRB review but do require WVU review and approval.
- For more information about the categories of exempt review, see 45 CFR Part 46, Protection of Human Subjects.
Limitations on Exemptions
Exemption for research involving survey or interview procedures or observations of public behavior does not apply to research in children, except for research involving observation of public behavior when the investigator does not participate in the activities being observed. Prisoner exemptions do not apply and required IRB review.
There are 18 HIPAA Identifiers that preclude an application from being an Exempt protocol. Review these identifiers and remove any that you may have planned to collect or your protocol will not qualify as an Exempt protocol.
- For more information about Exempt Research review the ORIC Exempt Research Checklist.
back to top ↑
Design

Figure 166 – Protocol Document, Questionnaire Page – Design section
Questions will be asked in the Design section regarding the design of the research study including observations or measures, treatments or programs, groups, assignment to groups, and timeframe. A lay terms description of the project should be a brief snapshot of the research proposal. Typical summaries might discuss the purpose of the research, its background significance, and the proposed methodologies. The purpose of this summary is to help the ORIC staff and IRB reviewers more easily understand the details of the proposed research. The summary must be simple and direct, explaining why the research is significant to the general public, be written at the 6th grade level, and include:
- A statement of the problem;
- statement of the purpose;
- A description of the instruments used in the study and any intervention;
- The number of participants in the study. Note that participant eligibility (i.e., who is allowed to participate) is more important than specific number;
- The length of the study;
- Specific study endpoints as well as potential beneficial and negative effects; and
- Notification to the subjects of the potential impact of the study.
The design is used to structure the research, to show how all of the major parts of the research project work together to address the central research question. Will the research be a randomized/true design, quasi-experimental design, or non-experimental design?
- If using surveys/interviews/questionnaires, a copy of the document MUST be attached on the Notes & Attachments page.
back to top ↑
Expedited Review
Quick Reference Guides
| Category | Requirements |
|---|---|
| Expedited Review Category 1 | Research on drugs for which an investigational new drug application is not required or research on FDA-approved medical devices. |
| Expedited Review Category 2 | Collection of blood samples by finger stick, heel stick, ear stick, or venipuncture. |
| Expedited Review Category 3 | Prospective collection of biological specimens for research purposes by noninvasive means (e.g., hair/nail clippings, teeth, excreta/external secretions, etc.). |
| Expedited Review Category 4 | Collection of data through noninvasive procedures routinely employed in clinical practice. Procedures do not include general anesthesia, sedation, x-rays, or microwaves. Studies intended to evaluate the safety and effectiveness of a medical device are not generally eligible. |
| Expedited Review Category 5 | Research involving materials that have been collected or will be collected solely for nonresearch purposes such as medical treatment or diagnosis. |
| Expedited Review Category 6 | Collection of data from voice, video, digital, or image recordings made for research purposes. |
| Expedited Review Category 7 | Research on individual or group characteristics or behavior or research employing survey, interview, oral history, focus group, program evaluation, human factors evaluation or quality assurance methodologies. |
| Expedited Review Category 8 | Continuing review of research previously approved by the convened IRB if one of the following conditions are met:
|
| Expedited Review Category 9 | Continuing review of research, not conducted under an investigational new drug application or investigational device exemption where Categories 2-8 do not apply but the IRB has determined and documented that the research involves no greater than minimal risk and no additional risks have been identified. |
Table 66 Protocol Document, Questionnaire Page – Full Board or Expedited Review Section, Expedited Review Categories
Expedited Review Limitations
The expedited review procedure may not be used where identification of subjects and/or their responses would reasonably place them at risk of criminal or civil liability or be damaging to the subjects financial standing, employability, insurability, reputation, or be stigmatizing, unless reasonable and appropriate protections will be implemented so that risks related to invasion of privacy and breach of confidentiality are no greater than minimal. Expedited review procedure may not be used for governmental classified research involving human subjects.
The activities listed below are eligible for review through the expedited review procedure when the specific circumstances of the proposed research involving no more than minimal risk to human subjects.
- Minimal risk indicates that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
- For more information about Expedited Reviews, see the ORIC Expedited Review Determination Checklist.
Intervention Research
Intervention includes both physical procedures by which data are gathered and manipulations of the subject or subject's environment that are performed for research purposes. If research is intervention research, indicate if the research can be categorized as medical research procedures.
Medical Research Procedures
The Guardianship and Administration Act 1986, defines medical research procedure as a procedure:
- Carried out for the purposes of medical research, including, as part of a clinical trial, the administration of medication or the use of equipment or a device;
- Prescribed by the regulations to be a medical research procedure for the purposes of the Act.
Medical research procedures DO NOT include:
- Any non-intrusive examination (including a visual examination of the mouth, throat, nasal cavity, eyes or ears or the measuring of a person’s weight, height, or vision); or,
- Observing a person’s activities; or,
- Undertaking a survey; or,
- Collecting or using information, including personal information or health information (regulated by other privacy and confidentiality laws); or,
- Any other procedure that is prescribed by the regulations not to be a medical research procedure for the purposes of the Act.
Risk Assessment
Identify the potential possibilities for risk or harm to the subjects as a result in participation in the research. Describe how the research team will interact with the study’s participants and how these interactions will be kept private.
- Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests
Cancer-Related Research
If the research deals with cancer prevention, treatment, or diagnosis, the protocol must be reviewed and approved by the Mary Babb Randolph Cancer Center’s Protocol Review and Monitoring Committee (PRMC). If the protocol has been reviewed and approved by the PRMC, attach the approval letter on the Notes & Attachments page. If the protocol has not been reviewed and approved by the PRMC, stop work on the protocol submission and contact the PRMC coordinator at 304-293-4944 immediately.
back to top ↑
Risk Assessment
Quick Reference Guides

Figure 167 Protocol Document, Questionnaire Page – Full Board or Expedited Review Section
Interventions
If using an intervention, a description must be given of the drugs or devices to be used, indicating if they are already commercially available or in phases of experimentation. For drugs and devices that are commercially available, the protocol must state their proprietary names, manufacturer, chemical composition, dose, and frequency of administration. For drugs and devices that are still in the experimental stage or that are commercially available but are being used for a different indication or in a different mode of administration, additional information should be provided on the results of studies already conducted on humans.
Medical Research Procedures
The Guardianship and Administration Act 1986, defines medical research procedure as a procedure:
- Carried out for the purposes of medical research, including, as part of a clinical trial, the administration of medication or the use of equipment or a device;
- Prescribed by the regulations to be a medical research procedure for the purposes of the Act.
Medical research procedures DO NOT include:
- Any non-intrusive examination (including a visual examination of the mouth, throat, nasal cavity, eyes or ears or the measuring of a person’s weight, height, or vision); or,
- Observing a person’s activities; or,
- Undertaking a survey; or,
- Collecting or using information, including personal information or health information (regulated by other privacy and confidentiality laws); or,
- Any other procedure that is prescribed by the regulations not to be a medical research procedure for the purposes of the Act.
Data and Safety Monitoring Board
A Data and Safety Monitoring Board (DSMB) is an independent group of experts that advises study investigators by providing expertise and recommendations. DSMBs periodically review and evaluate the accumulated study data for participant safety, study conduct, and progress as well as make recommendations concerning the continuation, modification, or termination of the study.
Data and Safety Monitoring Plan
A Data and Safety Monitoring Plan (DSMP), is meant to assure that each clinical investigation has a system for appropriate oversight and monitoring of the conduct of the clinical investigation. Such oversight ensures the safety of the participants and the validity and integrity of the data.
Risk Assessment
Identify the potential possibilities for risk or harm to the subjects as a result in participation in the research. Describe how the research team will interact with the study’s participants and how these interactions will be kept private.
- Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
Cancer-Related Research
If the research deals with cancer prevention, treatment, or diagnosis, the protocol must be reviewed and approved by the Mary Babb Randolph Cancer Center’s Protocol Review and Monitoring Committee (PRMC). If the protocol has been reviewed and approved by the PRMC, attach the approval letter on the Notes & Attachments page. If the protocol has not been reviewed and approved by the PRMC, stop work on the protocol submission and contact the PRMC coordinator at 304-293-4944 immediately.
back to top ↑
HIPAA
Figure 168 Protocol Document, Questionnaire Page – HIPAA Section
The Health Insurance Portability and Accountability Act of 1996 (HIPAA) created national standards for electronic healthcare transactions and protects the confidentiality of an individual’s protected health information (PHI). PHI is any health information that identifies an individual and is maintained or exchanged in hard copy or electronically. Protected PHI obtained by WVU may not be used internally or disclosed to any outside person or organization for research purposes without prior approval of the IRB. WVU researchers must also abide by all corporate HIPAA policies regarding HIPAA privacy and security.
In this section of the Questionnaire, indicate if the research involves PHI in any way. If so, indicate the PHI statement(s) that meets the research and attach the corresponding HIPAA form on the Notes & Attachments page.
- To access the HIPAA forms, see the ORIC Human Subject Research Forms webpage.
| HIPAA Forms | Description |
|---|---|
| HIPAA Form | Uploaded form must be .pdf. Authorization from the subject when the research being conducted involves PHI. |
| HIPAA Data Use Agreement Form | Uploaded form must be .pdf. Use when certain data elements that are not permitted in de-identified data are required for a study. A participant must disclose a limited data set to the investigator without authorization or waiver, provided that the investigator has signed a data use agreement. Such a limited data set is still considered to be protected health information (PHI), but it must exclude only specified direct identifiers. |
| HIPAA Decedents Form | Uploaded form must be .pdf. A participant may use or disclose the PHI of a deceased person (decedent) if the investigator indicates that the use or disclosure is solely for research on the PHI, that the PHI is being sought is necessary for the research, and that documentation of the death of the individual is being sought. |
| HIPAA De-Identification Certification | Uploaded form must be .pdf. Research that involves the use of de-identified PHI is exempt from HIPAA requirements. There are 18 identifiers that must be removed from the data set to create de-identified data. |
| HIPAA Research Authorization Form | Uploaded form must be .pdf. Written authorization to use, disclose, or share PHI with others for research purposes. Participants must sign this form in order to participate in a research study. |
| HIPAA Veterans Affairs Form | Uploaded form must be .pdf. Authorization from a veteran when the research being conducted involves PHI. For VAMC use only. |
| HIPAA Waiver of Research Authorization Form | Uploaded form must be .pdf. Use if the research cannot be practically done without a waiver (and not without access to and use of the PHI) and that disclosure poses minimal risk to privacy. |
Table 67 Protocol Document, Questionnaire Page – HIPAA Section, HIPAA Forms
Combined Forms
Indicate if you will be using a combined HIPAA and informed consent form.
- To use a combined consent and HIPAA form, save the form to your computer. Once the form has been edited as needed, save it as a PDF document. To do so, while the form is open in Microsoft Word, simply click the circular Windows button at the top left, click Save As, in the dialog box that appears select the “Save as type” PDF value, then click Save. If not presented with the option to save as a PDF, please email the Word form to Jonathan.Young@mail.wvu.edu and request a PDF copy in return.
- For more information about HIPAA, see the Summary of the HIPAA Privacy Rule.
back to top ↑
Subjects

Figure 171 Protocol Document, Questionnaire Page –Sample Size Section
Indicate the number of subjects that will be enrolled in the study and why that particular sample size was chosen. Provide information and justification about sample size. If this is a multi-site study, indicate the total number of subjects that will be participating in the study worldwide (including subjects at WVU).
- Determining sample size for a study is a crucial component of study design. A larger sample size than needed to test the research hypothesis increases the cost and duration of the study and will be unethical if it exposes human subjects to any potential unnecessary risk without additional benefit. A smaller sample size than needed can also be unethical if it exposes human subjects to risk with no benefit to scientific knowledge. Using the appropriate number of subjects optimizes the probability that a study will yield interpretable results and minimizes research waste.

Figure 170 Protocol Document, Questionnaire Page – Subjects Section
Identify and describe the types of subjects who will be participating in the research. Describe what will be done to find and recruit participants, indicating the gender(s) and age(s). For children under the age of 18, consent by a parent or guardian must be given. If not, a justification of the waiver of consent must be provided.
Vulnerable Populations
When some or all participants in a WVU research project are likely to be vulnerable to coercion or undue influence or have diminished decision-making capacity, the research must include additional safeguards to protect the rights and welfare of those participants. The Subjects section of the Questionnaire will allow users to outline how the general welfare of vulnerable populations will be protected (e.g., how informed consent will be obtained, how the subject's confidentiality will be protected, how undue coercion will be prevented). Vulnerable populations include:
| Vulnerable Populations | Description |
|---|---|
| Children | A child ages 29 days to 6 years. |
| Cognitively Impaired | A person’s ability to think, concentrate, formulate ideas, reason, and remembers is affected, either permanently or temporarily. This is distinct from a learning disability as it may have been acquired later in life as a result of an accident or illness. |
| Minors | A child ages 7 to 17 years of age. |
| Neonates | A newborn (0-28 days of age). |
| Pregnant Women & Fetuses | A woman is assumed to be pregnant if she exhibits any of the pertinent presumptive signs of pregnancy, such as missed menses, until the results of a pregnancy test are negative or until delivery. A fetus is the product of conception from implantation until delivery. After delivery, the placenta; the dead fetus, macerated fetal material; or cells, tissue, or organs excised from a dead fetus. |
| Prisoners | Any individual involuntarily confined or detained in a penal institution. |
| WVU/UHA/WVUH Employees | Employees of West Virginia University, University Health Associates, or WVU Healthcare. To minimize coercion, investigators should, whenever possible, avoid the use of their employees in procedures that are neither therapeutic nor diagnostic. The voluntary nature of their participation must be primary and without undue influence on their decision. |
| WVU Students | WVU students or students from any institution associated with the study. To minimize coercion, investigators should, whenever possible, avoid the use of their students in procedures that are neither therapeutic nor diagnostic. The voluntary nature of their participation must be primary and without undue influence on their decision |
Table 69 Protocol Document, Questionnaire Page – Subjects Section, Vulnerable Populations Descriptions
Exempt Studies
Describe how the research team will interact with the study’s participants and how these interactions will be kept private. Identify the potential possibilities for risk or harm to the subjects as a result in participation in the research.
- Minimal risk means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
back to top ↑
Potential Benefits
Figure 173 Protocol Document, Questionnaire Page –Potential Benefits Section
Describe the potential benefits to be gained by the individual as a result of participating in the study as well as the potential benefits to society and the scientific community.
Risk/Benefit Assessment
The IRB will assess the research to ensure that the risks to subjects posed by participation in the research are justified by the anticipated benefits to the subjects or society. This assessment will involve: identifying the risks associated with the research, determine whether the risks will be minimized to the extent possible, identify the probable benefits to be derived from the research, determine whether the risks are reasonable in relation to the benefits to subjects and assess the importance of the knowledge to be gained, and ensure that potential subjects will be provide with an accurate and fair description of the risks or discomforts and the anticipate benefits.
back to top ↑
Consent Procedures

Figure 174 Protocol Document, Questionnaire Page –Consent Procedures Section
No investigator conducting research at WVU may involve a human being as a subject in a non-exempt research study without obtaining the legally effective informed consent of the subject or the subject’s legally authorized representative unless a waiver of consent has been approved by the IRB. Informed consent is a vital part of the research process and entails more than obtaining a signature on a form. It is a process of information exchange between the prospective Human Subject and the Investigator to include reading and signing the informed consent document. Investigators must educate potential subjects to ensure that they can reach a truly informed decision about whether or not to participate in the research. A participant’s informed consent must be given freely, without coercion, and must be based on a clear understanding of what participation involves.
- To access the Consent Form templates, see the ORIC Human Subject Research Forms webpage. All consent forms must be uploaded as .pdf files. Please note that the forms on the ORIC website are Word documents, to allow for editing. Once you have edited the form with the necessary information, save the form as a .pdf file by clicking the circular Windows button at the top left, clicking Save As, selecting .pdf as the save as type, and then saving.
| Consent Form | Description |
|---|---|
| Children’s Assent Form | Assent is an agreement by an individual not competent to give legally valid informed consent (e.g., a child or cognitively impaired person) to participate in research. |
More than Minimal Risk |
Two characteristics influence the nature of the risk: (1) the probability of harm and (2) the magnitude of harm. The magnitude of harm can be related to the severity, duration, and reversibility of a potential harm. |
More Than Minimal Risk –Parental/Guardian |
Parental consent for a child under the age of 18 to participate in a study with more than minimal risk (i.e., the probability of harm or discomfort is greater than those encountered in daily life). |
Only Minimal Risk |
Minimal risk indicates that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests. |
Only Minimal Risk – Parental/Guardian |
Parental consent for a child under the age of 18 to participate in a study with only minimal risk (i.e., the probability of harm or discomfort is not greater than those encountered in daily life). |
| Short Form | A written document stating that the elements of informed consent required by regulation have been presented orally to the participant or the participant’s legally authorized representative. The short form consent document must be written in a language understandable to the participant or the participant’s legally authorized representative.
|
| Veterans Administration Consent Form | The legal agreement for a veteran to participate in a research study. For VAMC Use Only. |
Table70 Protocol Document, Questionnaire Page – Consent Procedures Section, Sample Consent Forms
Waiver of Informed Consent
A waiver of informed consent approves a consent procedure that does not include, or alters, some or all of the elements of informed consent or waives the requirements to obtain informed consent. If requesting a waiver of informed consent, a researcher must explain in the Consent Procedures section how waiving the requirements for informed consent will not adversely affect the rights and welfare of study subjects, that waiving the requirements for informed consent presents no more than minimal risk to study subjects, that the research could not be carried out practicably without the waiver, and any additional pertinent information that will be provided to the study subjects after participation.
Waiver of Documentation of Informed Consent
The IRB may waive the requirements for the investigator to obtain a signed consent form for some or all of the research subjects. If requesting a waiver of the requirement to obtain written documentation of the consent process, the investigator must provide a written summary of the information that will be communicated to the subjects, rationale explaining that the only record linking the subject and the research is the consent document and that the potential principle risk would be a breach of confidentiality, indicate how the research involves neither minimal risk to subjects nor any procedures for which written consent is normally required, and describe the proposed consent process.
Obtaining Consent
If someone other than the investigator conducts the interview and obtains consent from a subject, the investigator needs to formally delegate this responsibility to a person who has received appropriate training to perform this activity. Explain in the Consent Procedures section why it is necessary for someone other than the PI or Co-I to consent subjects and indicate how this person is knowledgeable about the research and the consenting process in order to answer questions about the study. List all other languages that the consent form will be translated into for any non-English speaking subjects participating in the study.
back to top ↑
Confidentiality
Figure 175 Protocol Document, Questionnaire Page –Confidentiality Section
In order not to use identifiable data, and be able to answer “No” to the first question in this section, data collection for the study must be anonymous. Anonymity can only be met if at no point in time the subjects can be identified by members of the research team.
Explain if there will be any way that that any member of the research team will be able to identify subjects using the study’s data and what steps will be taken to maintain confidentiality of such data in the Confidentiality section. If the data will be identifiable, the investigator must indicate who will have access to the data, how long it will be kept, where it will be securely located, and how it will be destroyed. Investigators must also identify the steps that will be taken to maintain the confidentiality of data and the privacy of the study’s subjects.
- Confidentiality and anonymity are not the same. If anyone, including an investigator, can readily ascertain the identity of the subjects from the data, then the research is not anonymous and the IRB must determine if appropriate protections are in place to minimize the likelihood that the information will be inappropriately divulged. The level of confidentiality protections should be commensurate with the potential of harm from inappropriate disclosure.
back to top ↑
Financial Considerations
Financial Considerations
Figure 176 Protocol Document, Questionnaire Page –Financial Considerations Section
Describe any costs to the subject that may result from participation in the research. Indicate who will cover the costs (e.g., sponsor, insurance, etc.), if there is any mechanism in place to help minimize or eliminate the cost to the subject, and if the subjects will be reimbursed for any costs incurred.
If payment will be made to subjects on this research study, indicate the total dollar amount or value (if gift certificates, coupons, etc.) of the payment and how the payments will be distributed to the subject in the Payments/Reimbursements section. Payment should accrue and not be contingent upon the participant completing the entire study.
- Payment to research subjects may be incentive for participation or a way to reimburse a subject for travel and other expenses incurred due to participation; however, payment for participation is not considered a research benefit. Investigators must take care to avoid coercion of subjects, but payments should reflect the degree of risk, inconvenience, or discomfort associated with participation.
Extra Credit for Students
If students are to receive class credit for participation, other opportunities must be available to earn equivalent credit. Only students from the Psychology Department, School of Journalism, or College of Business and Economics are approved to receive extra credit.
back to top ↑
Advertisements

Figure 178 Protocol Document, Questionnaire Page –Advertisements Section
Investigators will be able to add up to eight (8) advertisement methods for each study in the Advertisements section. Select all the advertisement methods being used from the drop-down  menu, list all the places the advertisement will be placed, and then attach a copy of the advertisement(s) on the Notes & Attachments page.
menu, list all the places the advertisement will be placed, and then attach a copy of the advertisement(s) on the Notes & Attachments page.
For studies being conducted under the WVU IRB purview, the IRB must approve any and all advertisements prior to being posted/distributed in order to assure that the material is accurate, is not coercive or unduly optimistic, or creates undue influence on the subject to participate. Advertisements should be limited to the information the prospective subjects need to determine their eligibility and interest. Use the WVU ORIC Advertisement Checklist to make sure that the advertisements for a research study meet all the necessary requirements. Once approved by the IRB, an advertisement cannot be altered or manipulated in any way without IRB approval.
back to top ↑
Drug/Substance/Nutrient/Biologic

Figure 179 Protocol Document, Questionnaire Page –Drug/Substance/Nutrient/Biologic Section
Investigators will be able to add information for up to three (3) drugs that are being used for investigational purposes only in the Drug/Substance/Nutrient/Biologic section. Indicate if the drugs are Investigational New Drugs (IND) and where the drugs will be stored. For all INDs, investigators must provide the IND number, describe the side effects of the drug (specifically the serious and/or life-threatening), and any other pertinent information related to the drug. Non-investigational drugs that are being used in the study will also need to be listed along with the off-label use.
Investigational Drugs
An Investigational New Drug (IND) is a drug that has not been approved for general use by the FDA but is under investigation in clinical trials regarding its safety and efficacy first by clinical investigators and then by practicing physicians using subjects who have given informed consent to participate. Investigators must indicate if the use of an IND for the proposed research study is anticipated and provide documented assurance from the sponsor that the manufacture and formulation of the investigational drug conforms to federal regulations. Documentation of the IND could be an industry-sponsored protocol with IND, a letter from the FDA, a letter from the industry sponsor, or another document/communication verifying the IND. If the research involves drugs and there is no IND, the investigator must provide rationale why it is not required.
- An investigator’s brochure MUST be attached on the Notes & Attachments page for all INDs.
IND Exemptions
An IND is not necessary if the drug being used in the research is lawfully marketed in the United States and meets all of the following requirements:
- The research is not intended to be reported to the FDA in support of a new indication for use or to support any other significant change in the labeling of the drug;
- The research is not intended to support a significant change in the advertising for the product;
- The research does not involve a route of administration or dosage level, use in a subject population, or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product;
- The research is conducted in compliance with the requirements for IRB review and informed consent;
- The research is conducted in compliance with the requirements concerning the promotion and sale of drugs;
- The research does not intend to invoke FDA regulations for planned emergency research.
WVU Healthcare Pharmacy
If proposing to use a drug in research, investigators must provide a plan that includes storage, security, and dispensing of such drug/substance/nutrient/biologic. All drug storage must be approved by the WVU Healthcare Pharmacy. If using a controlled substance, the item should be ordered and received by WVUHP and then re-issued in appropriate quantities to researchers for human studies, pursuant to a study-specific and patient-specific medication order developed by WVUHP in collaboration with the investigator.
back to top ↑
Device Information

Figure 180 Protocol Document, Questionnaire Page –Device Information Section
Investigators will be able to add information for the devices that are being used in the study in the Device Information section Indicate if the device is Class IIB, approved by FDA for marketing, is being used in an off-label indication, is an implant, if it supports/sustains human life, is important in diagnosing/curing/mitigating/treating disease, or is an investigational device. If using an investigational device, the investigator must provide the IDE number, a risk assessment of the device, and his/her qualifications for using the device.
Class II Devices
Class II devices are those for which general controls alone cannot assure safety and effectiveness and are also subject to special controls which could include special labeling requirements, mandatory performance standards and postmarket surveillance.
Investigational Devices
Investigational Devices are any healthcare product that does not achieve its primary intended purposes by chemical action or by being metabolized. A medical device that is the subject of a clinical study is designed to evaluate the effectiveness and/or safety of the device. Investigators must indicate if the use of an investigational device for the proposed research study is anticipated. If so, the investigator must indicate if there is an investigational device exemption (IDE) and provide documented assurance from the sponsor that the manufacture and formulation of the investigational device conforms to federal regulations. Documentation of the IDE could be an industry-sponsored protocol with IDE, a letter from the FDA, a letter from the industry sponsor, or another document/communication verifying the IDE. If the research involves a device and there is no IDE, the investigator must provide rationale why it is not required.
Humanitarian Use Device Status
The Humanitarian Use Device (HUD) program creates an alternative pathway for getting market approval for medical devices that may help people with rare diseases or conditions. A HUD is a medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the U.S. per year. See the Humanitarian Use Devices section, for more information about submitting a HUD protocol.
- For more information, see Class II Special Controls Guidance, FAQs about IDE, and Designating Humanitarian Use Devices on the FDA website.
back to top ↑
Sample Collection
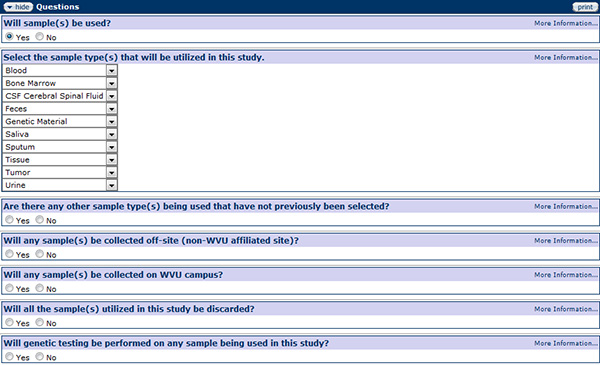
Figure 181 Protocol Document, Questionnaire Page – Sample Collection Section
Tissue samples include any cell tissue, fluid, or excreta from which measures of normal or pathologic human physiologic function can be obtained such as hair and nail clippings, deciduous and permanent teeth, dental plaque and calculus, sweat, uncannulated saliva, placenta removed at delivery, amniotic fluid, cerebrospinal fluid, genetic material, urine, blood, and other bodily fluids. If collecting samples, investigators must indicate in the Sample Collection section the various types of samples that will be collected, the purpose of the collection, and indicate if the samples will be shipped to another location or discarded. If being shipped to another location, the investigator must have completed the Shipping and Transport of Regulated Biological Materials training. If the samples will be provided by an external source, the investigator must provide the source of the collection and indicated if the samples are de-identified. If genetic testing is to be performed on the sample, indicate if the test results will be given, if disease risk will be quantified, if change in a family relationship will be disclosed, or if the subject/family member has the option not to know the results, and explain how such a decision will be recorded. Additionally, indicate if there is any other clinically relevant information that could be uncovered by the study and how the disclosure of this added information will occur.
- Investigators collecting biological specimens must obtain approval from the Institutional Biosafety Committee (IBC) to conduct research. After obtaining IBC approval, attach the approval letter to the protocol on the Notes & Attachments page. The IRB will not review a protocol without an IBC approval letter.
- For more information about research biosafety, see the ORIC website.
Tissue Banking
If any tissue collected as part of an approved protocol will be banked for future research investigations at WVU, the investigator must first register the tissue bank and then conform to the guidelines set forth in the WVU Specimen Bank Policy. Investigators intending to create an on-site database or tissue bank utilizing clinical health information or authorized tissue specimens must complete and submit to ORIC an Application to Establish an On-Site WVU Tissue Bank for Research or Database Development Involving Specimens Stored With or Without Clinical Health Information form. An approved tissue-banking form should be submitted as part of the protocol application.
back to top ↑
Radiation Safety
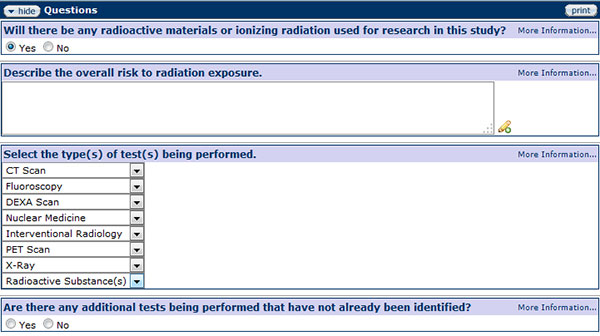
Figure 182 Protocol Document, Questionnaire Page –Radiation Safety Section
If using radioactive materials or ionizing radiation in a study, please describe the overall risk to exposure, select the type of test being performed, describe the body area being exposed, and explain the exposures specifically for research purposes in the Radiation Safety section.
- All research activities involving the use of radioactive materials, radiation producing devices and the diagnostic and therapeutic use of radiation in humans, non-humans, and animals is overseen by the WVU Radiation Safety Department and its subcommittees. The Radiation Safety Department will provide guidance and enforcement to guarantee a safe working environment for all individuals working with radioactive materials or devices located within the WVU Campuses, the Robert C. Byrd Health Sciences Center, Jefferson Memorial Hospital, WVU Hospitals, Inc., and Blanchette Rockefeller Neurosciences Institute.
back to top ↑
Biological Safety

Figure 183 Protocol Document, Questionnaire Page – Biological Safety Section
Investigators should indicate in the Biological Safety section if infectious agents and/or non-infectious agents or recombinant DNA is being handled through the research.
- The Institutional Biosafety Committee (IBC) oversees all activities that pose a biohazard. IBC approval is required for human research activities involving infectious agents, the use of serum and/or tissue, creation of transgenic eukaryotes, or the transfection using adenovirus-derived vectors or other vectors capable of infecting human cells. The IBC approval letter must be attached on the Notes & Attachments page before the protocol can be submitted.
- For more information about research biosafety, see the ORIC website.
back to top ↑
Emergency Use
Quick Reference Guides
Figure 342 Protocol Document, Questionnaire Page – Emergency Use Section
Emergency Use is the use of an investigational drug or biological product with a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval. The IRB must be notified within five (5) working days when an emergency exemption is used. Any subsequent use of the test article at WVU is subject to IRB review. This notification should not be construed as an approval for the emergency use by the IRB. The IRB will review the report to verify that circumstances of the emergency waiver conform to FDA regulations.
Indicate the type of test article used/planned to be used, the name of the test article, and date the test article was used/is planned to be used. Informed consent must be obtained and documented in writing.
- A copy of the consent form used must be attached on the Notes & Attachments page.
Waiver of Informed Consent
Informed consent must be obtained and documented in writing unless all of the following exemption criteria can be met and verified by an independent physician:
- The subject is confronted by a life-threatening situation necessitating the use of the test article;
- Informed consent cannot be obtained because of an inability to communicate with, or obtain legally effective consent from, the subject; and,
- Time is not sufficient to obtain consent from the subject’s legally authorized representative;
- No alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the subject’s life.
If time is not sufficient to obtain the independent physician determination before use of the test article, the actions of the investigator must be reviewed and evaluated in writing by an independent physician within 5-6 working days.
- Explanation from an independent physician why informed consent was not obtained MUST be attached on the Notes & Attachments page.
back to top ↑
Humanitarian Use Devices
Quick Reference Guides
A Humanitarian Use Device (HUD) is a device that is intended to be used in fewer than 4,000 individuals in the U.S. per year. Treatment with a HUD is subject to full board initial review and continuing review by the IRB. In order for a HUD to be used in treatment, diagnosis, or research at WVU, the IRB and FDA must approve it and a Humanitarian Device Exemption (HDE) must be issued.
Cover Letter
In order to obtain an HDE determination, the patient must be informed of the HUD via a cover letter. Informed consent is not required since the use is not considered research. The cover letter must be attached to the HUD protocol and include the following:
- Explain a HUD and what it means;
- List all risks and procedures associated with the use of the HUD;
- Inform the patients that the device is approved only for certain uses and that the patient meets the conditions of the intended use; and,
- Inform the patient that the costs will be charged to the patient or the patient's insurance.
HUD Renewal
When submitting a renewal for a HUD protocol, a document must be attached addressing the following information:
- Number of times the HUD has been used, listed chronologically;
- Copy of the hospital consent indicating the use is for HUD;
- Copy of the note in the patient's chart stating that the patient received the cover letter;
- Any problems associated with its use; and,
- Any information received from the sponsor or the FDA concering the use of the HUD at other sites.
UPIRTSOs
If a PI receives or becomes aware of information that a HUD has or may have caused or contributed to the death or serious injury of a patient, such findings must be reported to the FDA and IRB as soon as possible but no later than ten (10) working days.
Emergency Use of a HUD
During an emergency, if IRB approval cannot be obtained in time to prevent serious harm or death to a patient, a HUD may be administered without prior IRB approval. In such an instance, approval must be obtained from the Chief of Staff and the PI is required to provide written notification of the use to the IRB within five (5) days after use of device via an Emergency Use Protocol.
back to top ↑